人类生殖细胞突变背后的生物学机制很大程度上并不清楚,近几十年的实验研究揭示出了各种各样DNA复制和修复的可能错误模式,但是还没有哪种机制被阐明可能是人类生殖细胞突变的主要原因。为了对人类生殖细胞系中的突变进行全面分类和理解,美国哈佛医学院Shamil Sunyaev研究组在Science发文题为Population sequencing data reveal a compendium of mutational processes in the human germ line,通过使用体积正则化非负矩阵分解(Volume-regularized nonnegative matrix factorization)的方法,给出了群体测序数据统计分解沿基因组突变率和图谱的变异过程,揭示出人类生殖细胞突变过程的概要。

在该工作中,作者们所采用的思路是利用基因组的突变率变化来模拟生殖细胞系的突变。作者们通过对基因组中192个三核苷酸突变类型和不同强度的谱图来对生殖细胞系中的突变机制进行描述。从基因组位点突变图谱的变异性推断突变过程其实是一个经典的非负矩阵因子分解(Nonnegative matrix factorization,NMF)的问题。NMF将一组非负信号也就是突变过程从观察到的突变频率中分离出来。然而,NMF可以有许多具有相同近似性质的解,所以在该问题单纯的NMF并不能对生殖细胞系中的突变机制进行解释。
所以作者们对NMF的方法进行了优化,构建了体积正则化非负矩阵分解的方法对生殖细胞系中的突变与突变频率之间的机制进行分类和揭示。反向平行DNA链之间的对称性为评估和推断生物学过程相关性提供了一种有力的方法。转录和复制等过程会打破这种对称性,而这些机制与突变机制是相互依赖的。对于一个DNA链依赖的突变机制,统计程度相同,举例来说A>G与T>C的突变频率是相同的,但是对于DNA链非依赖的突变机制这两种突变的频率则并不相同。作者们将该体积正则化非负矩阵分解的方法应用到TOPMed 数据库中2.92亿非常罕见的单核苷酸变异的分析之中。对该数据库应用体积正则化非负矩阵分解后作者们共识别出14种突变以及突变频率分类(图1),并且通过对gnomAD数据库的重采样分析发现具有同样稳健性和准确性的组成分类。
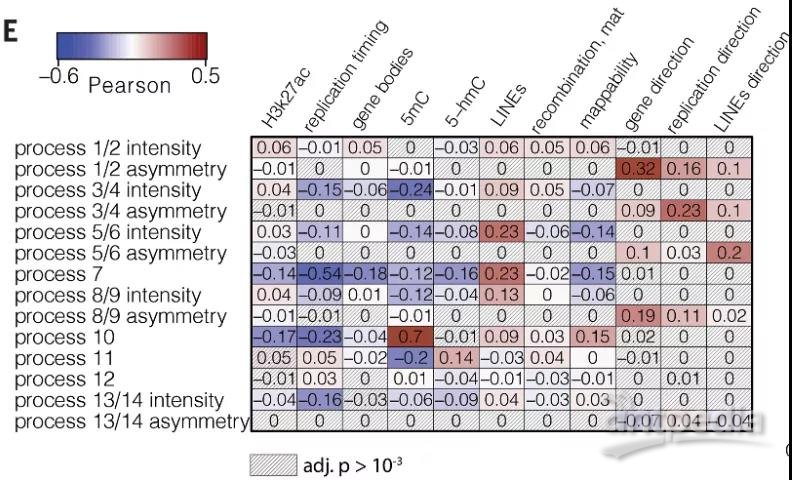
图1 体积正则化非负矩阵分解TOPMed数据库的14个组分分类以及相应突变率
14个组分分别对应9个突变过程,其中五个DNA链依赖的由两种组成分类表示以及4个DNA链独立的突变过程。这些过程只与一个或两个基因组特征相关,包括基因体、复制时间、复制方向和染色质可及性。举例来说,过程1/2是DNA链依赖的,是大体积DNA损伤不对称分辨的足迹,在发育早期的强度较低,嵌合A>G/T>C突变转录对称的方向相反。DNA链依赖过程3/4捕获的是不对称复制错误,其不对称性与复制的方向有关,由大量的DNA损伤组成的。过程5/6在L1PA LINE重复序列的非转录链上强度升高。再比如,过程10是以CpG转变为特征,主要由甲基胞嘧啶脱氨或甲基胞嘧啶的错误复制所介导的。
总的来说,作者们的工作通过体积正则化非负矩阵分解的方法揭示出了TOPMed数据库中突变特性差异的发生过程,并且作者们为其中的多个过程提供了可能的生物学机制解释,为理解人类生殖细胞系中突变的发生过程给出了新的分析方法和思考方式。
原文链接:
http://doi.org/10.1126/science.aba7408
来源: “BioArt” 如有侵权请联系删除 !




