多囊肾,即正常肾组织被无数小囊所代替,外形似一串葡萄。可分为4型:①常染色体隐性多囊肾;②肾发育不良;③常染色体显性多囊肾;④尿道梗阻多囊肾。其中常染色体显性多囊肾又称成人型多囊肾(ADPKD),发病率为1/500~1000。临床特征包括充满液体的囊肿的生长,使肾脏变形和扩大,并引发炎症和纤维化等继发性过程,这导致大多数患者出现肾功能衰竭。
现已证实,ADPKD是一种常见的遗传性疾病,主要由PKD1和PKD2突变引起,PKD1和PKD2分别编码polycsytin-1 (PC1)和polycytin -2 (PC2)。在ADPKD中,当肾小管细胞因体细胞"二次打击"突变而导致PKD1或PKD2无效时,囊肿就开始形成。随后囊肿进展,通过肾小管细胞形状、增殖和分泌的变化重塑器官使肾脏出现炎症和纤维化。目前基于同源基因Pkd1或Pkd2的小鼠模型有助于理解ADPKD的体内机制,并已用于临床前评估治疗。但减缓囊肿进展并不是长久之计,是否能在体内生理水平上实现致病PKD基因功能性再表达的逆转才是根本之策。
2021年10月11日,美国耶鲁大学StefanSomlo研究小组在《自然—遗传学》杂志上在线发表了一篇标题为“Renal plasticity revealed through reversal of polycystic kidneydisease in mice”的文章,研究通过逆转小鼠的多囊肾疾病揭示肾脏的可塑性。

为了确定ADPKD是否可以通过在体内生理水平上致病Pkd基因的功能性再表达逆转这个问题,研究团队开发了一种Pkd2失活和随后再激活的小鼠模型,来检测Pkd2连续失活后再激活对肾脏结构和功能的影响。实验表明,当诱导Pkd2失活后,小鼠未表达Pkd2编码的PC2-HA蛋白,并且小鼠在16周龄时肾脏重量:体重(KW:BW)比、矢状横截面囊性面积百分比(囊性指数,CI)和血尿素氮(BUN)显著增加。而在激活Pkd2后,在整个肾脏中可观察到PC2-HA的表达,从13周开始重新表达PC2-HA的小鼠也具有正常的KW:BW比率、CI和BUN。此外,研究人员采用MRI技术对模型小鼠的PC2-HA表达进行了体内监测,发现PC2-HA的缺失和再表达前后肾脏的组织学和体积均有明显的不同。
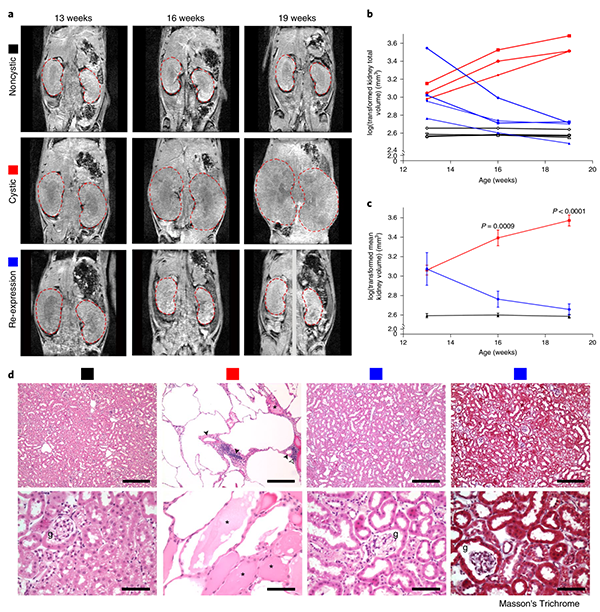
(图注:MRI和组织学检查观察PC2再表达后肾脏的变化)
随后,研究人员对囊肿中Pkd2活化后的现象进行了深入的机制研究。研究表明,PC2-HA的再表达可使肾单位逐渐恢复立方结构,并逆转管腔扩张,表明肾脏可塑性的发生可通过调节小管及其组成细胞的结构来实现。而在PC2激活后的快速组织重塑中,自噬也发挥了作用,同时PC2-HA的再表达导致ADPKD囊肿内壁细胞增殖的快速和完全逆转,而这种细胞增殖作用可能与Cyclin-D1相关(Cyclin-D1的功能是促进细胞增殖)。
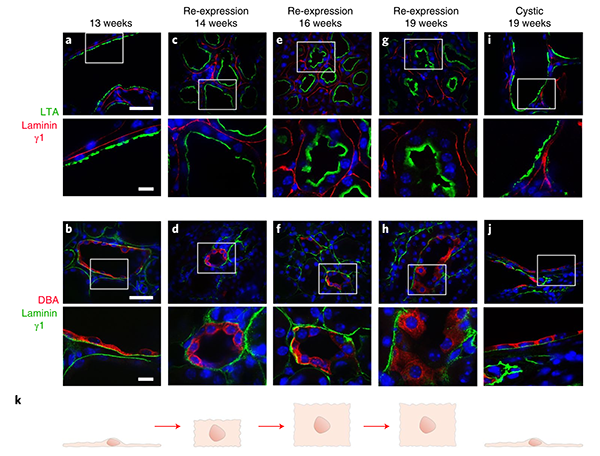
(图注:PC2再表达后小管细胞形态的变化)
最后,研究团队分析了PC2再表达后对炎症、肾脏小管间质纤维化的影响,发现炎症、细胞外基质沉积和肌成纤维细胞激活的增加可以被成功逆转,而且肾脏变得更小。并且检测了PC2再激活在ADPKD更晚期的小鼠中的作用,发现小鼠肾脏大小和CI可以恢复正常,但BUN没有恢复到正常,这可能是因为潜在的瘢痕、纤维化和肾元流失是不可逆的。
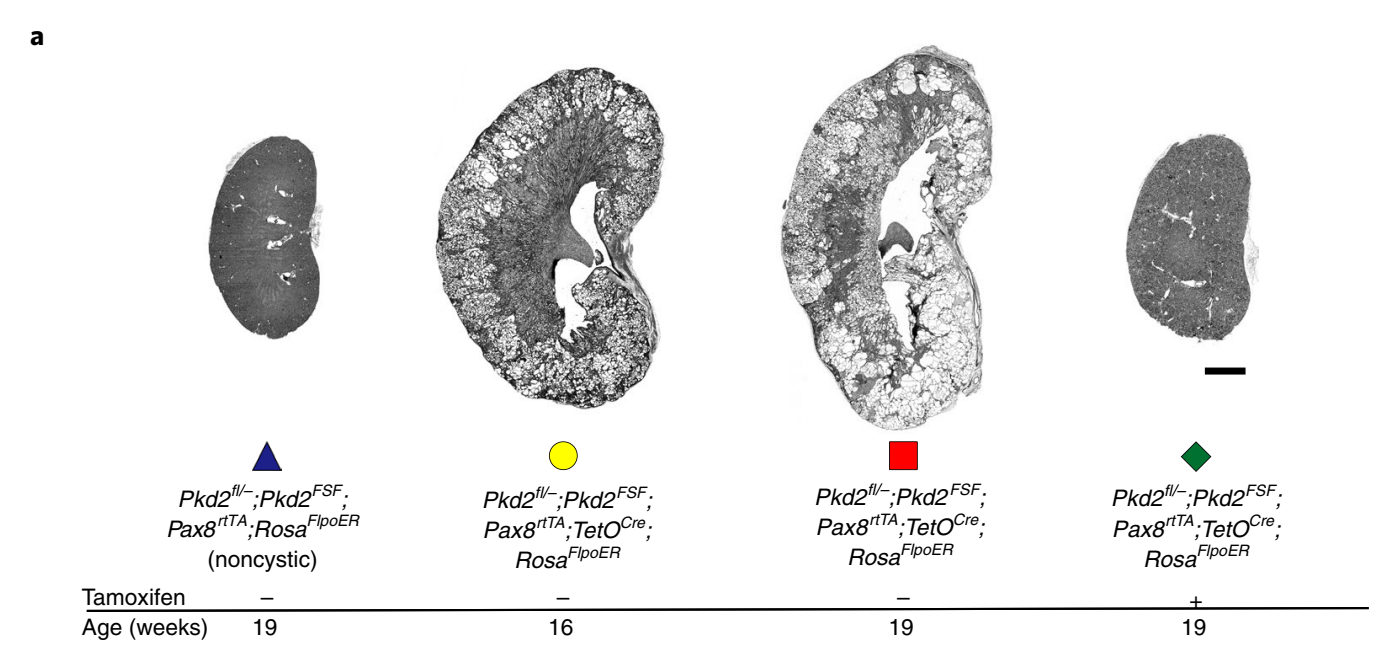
(图注:晚期Pkd2再激活可逆转囊肿的形成,但肾脏修复不完全)
总之,研究人员构建了一个小鼠模型,发现Pkd基因在囊性肾脏中的重新表达可导致ADPKD的快速逆转。囊肿细胞增殖减少,自噬作用被激活,由鳞状细胞内衬的管腔扩大的囊肿小管恢复到由立方体细胞内衬的正常管腔。可以从中得到结论是,ADPKD的表型特征是可逆的,肾脏有一种意想不到的可塑性能力,至少部分受ADPKD基因功能控制。
参考资料:
[1]Renal plasticity revealed through reversal of polycystic kidney disease in mice
转自网络如有侵权请联系删除 !




